




Abstract
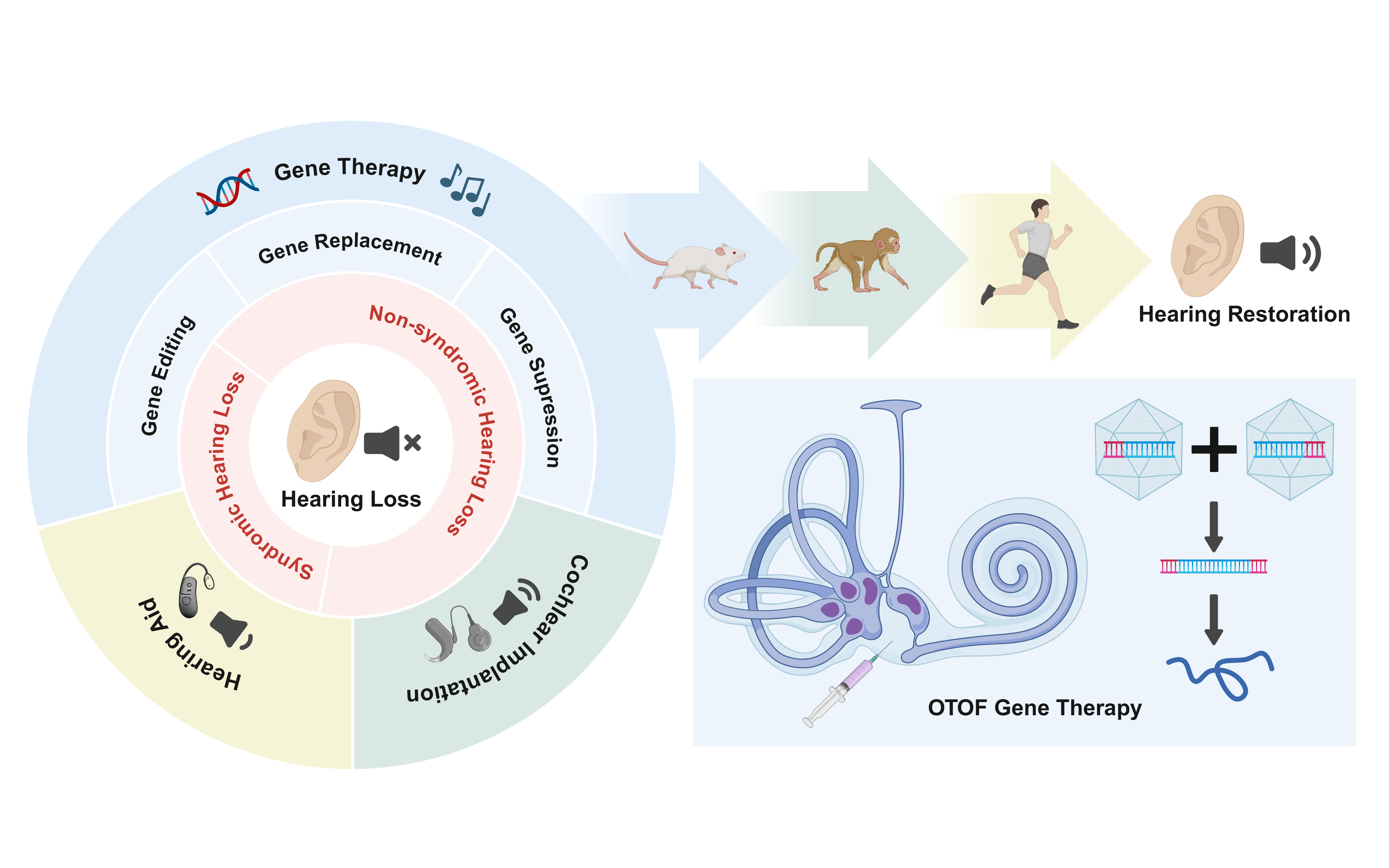
Hereditary hearing loss accounts for over 60% of congenital deafness cases, with non-syndromic hearing loss (NSHL) representing the most common subtype. Typically caused by monogenic mutations, NSHL presents a promising candidate for gene therapy. Recent advances in deciphering the genetic underpinnings of deafness and developing gene delivery systems have greatly accelerated the progress of inner ear gene therapy, leading to a number of breakthrough achievements. This review provides an overview of the latest developments in gene therapy for NSHL. After outlining the genetic basis of NSHL, we summarize the preclinical progress made during the first decade of hereditary deafness gene therapy. Special emphasis is placed on gene replacement strategies for DFNB9, an autosomal recessive form of hearing loss caused by mutations in the OTOF gene. We highlight the remarkable journey of OTOF gene therapy and discuss future directions in this transformative field.
Keywords: non-syndromic hearing loss; gene therapy; OTOF; DFNB9.
Introduction
Hearing loss is projected to affect 2.45 billion individuals by 2050, with consequences extending far beyond health impairments [1, 2]. Over 60% of congenital and childhood hearing loss cases are hereditary, which is further categorized into syndromic hearing loss and non-syndromic hearing loss (NSHL) [3, 4]. Accounting for approximately 70% of hereditary hearing loss cases, NSHL represents a major clinical entity. It primarily involves the auditory system and is predominantly monogenic, with well-characterized genetic mechanisms [5]. To date, 156 genes associated with NSHL have been identified, yet no pharmacotherapeutic options are commercially available.
Currently, cochlear implantation remains the clinical gold standard for hearing loss intervention. This device bypasses damaged hair cells by directly stimulating the auditory nerve with electrical pulses to transmit sound signals to the brain [6]. However, it does not correct the underlying genetic defects in target cells, nor does it restore natural physiological hearing [7, 8].
Gene therapy, a concept first proposed nearly six decades ago, is now recognized as a promising treatment for a wide range of genetic disorders [9]. It involves the manipulation of gene presence or expression and is generally implemented through three main strategies: gene replacement, gene suppression, and gene editing [10]. More than 3,000 clinical trials targeting genetic diseases across multiple organ systems are currently underway [11]. Patients with various conditions, including blindness, neuromuscular disorders, hemophilia, primary immunodeficiencies, and certain cancers, have already derived clinical benefits from gene therapy interventions. Given its typically monogenic etiology and confined anatomical focus, NSHL is considered an ideal candidate for inner ear gene therapy. Combined with next-generation sequencing based diagnostics that enable precise genetic characterization, this approach holds the potential to directly restore auditory cell function, offering a curative solution.
This rapidly evolving field has generated substantial progress. Successful translation from animal studies to clinical trials has also brought hope to patients with hereditary deafness [12-16]. Notably, a comparative study suggests that gene therapy may present several advantages over cochlear implants, including faster recovery of auditory function, improved speech perception in noisy environments, and better music discrimination [17]. Given limitations such as short follow-up periods and small sample sizes, it would be premature to conclude which approach is superior. However, these findings indicate that gene therapy may provide a novel effective treatment for genetically driven congenital deafness.
In light of the remarkable advances in deafness gene therapy in recent years, a timely synthesis of these developments is essential to consolidate knowledge and guide future research. This review summarizes the latest progress in gene therapy for NSHL and discusses its clinical implications. We place particular emphasis on gene replacement strategies for DFNB9, an area that has attracted extensive research interest and exhibits strong translational potential.
The Genetic Basis of non-syndromic hereditary hearing loss
NSHL exhibits heterogeneous inheritance patterns, predominantly autosomal recessive (75-80%) and autosomal dominant (about 20%), with minor contributions from sex-linked (about 2%) and mitochondrial (less than 1%) patterns[18, 19]. To date, 156 causative genes have been identified in major databases such as ClinVar, OMIM, and the Hereditary Hearing Loss database [5, 20], providing a foundational framework for the understanding of NSHL pathogenesis. Among these, GJB2 and SLC26A4 are the most frequently implicated pathogenic genes, with other significant contributors including MT-RNR1, OTOF, MYO15A, MYO7A, and TMC1, among others [21-24]. Allelic heterogeneity further compounds this genetic complexity, wherein multiple distinct deafness-causing mutations can occur within a single gene [25].
Autosomal recessive NSHLCurrently, 88 genes have been identified to cause autosomal recessive NSHL, the most prevalent subtype of NSHL [5]. Over 90% of autosomal recessive NSHL cases manifest as prelingual, severe-to-profound sensorineural hearing impairment, which typically affects all frequencies. This clinical presentation can be attributed to the critical roles played by the corresponding proteins in the development, structural integrity, and physiological function of various inner ear structures, including stereocilia bundles, the mechanoelectrical transduction (MET) channel complex, and the stria vascularis (Table 1).
Among these causative genes, GJB2 is the most prevalent, accounting for approximately half of congenital SNHL cases worldwide [26]. It encodes connexin 26, a gap junction protein essential for intercellular permeability [27]. SLC26A4 represents another common deafness gene, which is involved in anion transport, and mutations in this gene are associated with an enlarged vestibular aqueduct [28]. Another significant gene is OTOF, which has emerged as a promising target for gene therapy. It encodes otoferlin, a calcium sensor believed to regulate exocytosis and highly expressed in inner hair cells [29].
Table 1. Autosomal recessive NSHL
| Locus name | Gene symbol | Role in the inner ear |
|---|---|---|
| DFNB1A | GJB2 | Physiological ion balances |
| DFNB3 | MYO15A | Stereocilia elongation |
| DFNB4 | SLC26A4 | Anion and bases transmembrane transport |
| DFNB6 | TMIE | MET complex component |
| DFNB7/11 | TMC1 | MET complex component |
| DFNB8/10 | TMPRSS3 | Signaling regulation through proteolytic activation |
| DFNB9 | OTOF | Ca2+ sensor for exocytosis in hair cells |
| DFNB12 | CDH23 | Adhesion protein assisting stereocilia organization |
| DFNB15/72/95 | GIPC3 | Vesicle trafficking protein complex component |
| DFNB16 | STRC | Stereocilia cohesion, apical tip positioning |
| DFNB18B | OTOG | Sensory epithelial patches component |
| DFNB21 | TECTA | Hardesty's membrane component |
| DFNB22 | OTOA | Anchoring protein of tectorial membrane |
| DFNB24 | RDX | Cross-linkers between integral membrane proteins and actin of cytoskeleton |
| DFNB25 | GRXCR1 | Modulator of actin cytoskeleton in stereocilia development |
| DFNB28 | TRIOBP | Actin-binding Protein |
| DFNB29 | CLDN14 | Selective paracellular permeability |
| DFNB30 | MYO3A | Stereocilia component |
| DFNB31 | WHRN | Stereocilia elongation |
| DFNB32/105 | CDC14A | Mitosis assistance |
| DFNB35 | ESRRB | Development of marginal cells and stria vascularis |
| DFNB36 | ESPN | Myosin III cargo protein |
| DFNB39 | HGF | Regulation of epithelial cell development and motility |
| DFNB42 | ILDR1 | Integral protein of the tricellular tight junction complex |
| DFNB48 | CIB2 | Intracellular calcium signaling mediation |
| DFNB49 | MARVELD2 | Integral membrane protein in tight junction strand |
| DFNB53 | COL11A2 | Tectorial membrane component |
| DFNB57 | PDZD7 | Hair-cell stereocilia ankle-link complex composition |
| DFNB59 | PJVK | Stereocilia maintenance, pexophagy against the oxidative stress |
| DFNB63 | LRTOMT | TMC1/2 localization to the MET complex |
| DFNB67 | LHFPL5 | MET complex component |
| DFNB68 | S1PR2 | S1P-mediated cellular response and calcium signaling assistance |
| DFNB73 | BSND | Accessory subunit of chloride channels |
| DFNB74 | MSRB3 | Oxidatively damaged protein repairment |
| DFNB76 | SYNE4 | Intracellular organelle positioning |
| DFNB77 | LOXHD1 | Stereociliary bundle stabilization |
| DFNB79 | TPRN | Stereocilia taper composition |
| DFNB84A | PTPRQ | Phosphoinositide-mediated cellular regulation |
| DFNB84B | OTOGL | Acellular structures production or function |
| DFNB86 | TBC1D24 | Proper intracellular vesicle trafficking maintenance |
| DFNB91 | SERPINB6 | Cochlear homeostasis preservation |
| DFNB93 | CABP2 | Calcium signaling pathway modulation |
| DFNB101 | GRXCR2 | Cochlear stereocilia bundle integrity maintenance |
| DFNB102 | EPS8 | Stereocilia elongation, EGFR signaling and trafficking |
| DFNB104 | RIPOR2 | Circumferential ring near basal tapers of stereocilia |
| DFNB106 | EPS8L2 | Actin remodeling in response to EGF stimulation |
| DFNB111 | MPZL2 | Homophilic intercellular adhesion promotion |
| DFNB113 | CEACAM16 | Connection between stereocilia and Hardesty's membrane |
To date, 64 genes have been identified as causative for autoso-mal dominant NSHL [5]. The products of these genes are primar-ily involved in the maintenance and homeostatic regulation of inner ear structures, including cellular activity, functional stability, and transcriptional control (Table 2). Consequently, the associated clinical phenotype is typically less profound than that of auto-somal recessive NSHL. It is generally characterized by post-lin-gual onset, emerging from late childhood to early adulthood, and featuring progressive deterioration. This form of hearing loss is usually bilateral, predominantly affects high frequencies (resulting in a sloping audiometric configuration), and varies in severity from mild to profound [19, 30].
Common forms of autosomal dominant NSHL involve genes such as KCNQ4, TECTA, POU4F3, WFS1, EYA4, and ACTG1 [30-32]. Notably, some cases exhibit gene-specific frequency impairments. For example, mutations in DIAPH1 (DFNA1) and WFS1 (DFNA6/14/38) typically affect low frequencies, while DFNA16 (for which the causative gene remains unidentified) is characterized by fluctuating hearing loss [30].
Table 2. Autosomal dominant NSHL
| Locus name | Gene symbol | Role in the inner ear |
|---|---|---|
| DFNA1 | DIAPH1 | Cytoskeletal organization regulation |
| DFNA2A | KCNQ4 | Cellular repolarization |
| DFNA4A | MYH14 | Actin-cytoskeleton interactions, cytokinesis, motility and polarity regulation |
| DFNA4B | CEACAM16 | Connection between stereocilia and Hardesty's membrane |
| DFNA5 | GSDME | Pyroptosis assistance |
| DFNA6/14/38 | WFS1 | ER calcium channel or ER calcium channel regulator |
| DFNA7 | LMX1A | Neural progenitor specification and dopamine neurogenesis promotion |
| DFNA8/12 | TECTA | Tectorial membrane component |
| DFNA9 | COCH | Innate immunity |
| DFNA10 | EYA4 | Innate immune response cotranscription factor |
| DFNA11 | MYO7A | MET complex component |
| DFNA13 | COL11A2 | Tectorial membrane component |
| DFNA15 | POU4F3 | Transcription factor related to hair cells maintenance |
| DFNA17 | MYH9 | Actin-cytoskeleton interactions, cytokinesis, motility and polarity regulation |
| DFNA20/26 | ACTG1 | Structural and functional maintenance of hair cells |
| DFNA22 | MYO6 | Intracellular vesicle and organelle transport |
| DFNA25 | SLC17A8 | Glutamate synaptic vesicle transport |
| DFNA28 | GRHL2 | Epithelial morphogenesis and epidermal development promoter |
| DFNA36 | TMC1 | MET complex component |
| DFNA37 | COL11A1 | Tectorial membrane component |
| DFNA41 | P2RX2 | Temporary threshold shift for cochlea protection |
| DFNA50 | MIR96 | mRNAs translation and stability assistance |
| DFNA66 | CD164 | Cell adhesion receptor |
| DFNA67 | OSBPL2 | Lipid metabolism (possibly) |
| DFNA68 | HOMER2 | Stereociliary scaffolding protein |
| DFNA82 | ATP2B2 | Stereociliary calcium remove |
| DFNA84 | ATP11A | Phospholipid transport and uphill ion transport across membranes execution |
Only a negligible proportion of NSHL cases are attributed to mutations in sex chromosomes or mitochondrial DNA [18]. Sex-linked hereditary hearing loss demonstrates a distinct gender bias in its inheritance pattern, characterized by a higher prevalence, earlier onset, and more severe impairment in males than in females [33], [5, 34]. To date, seven DFNX and two DFNY loci have been identified as contributors to this form of hearing impairment. The Clinical Genome Resource (ClinGen) has established definitive gene-disease relationships for NSHL involving POU3F4, PRPS1, SMPX, and AIFM1 [35]. Among these, POU3F4 (DFNX3) is the most frequently implicated X-linked locus [34].
Mitochondrial NSHL is a maternally inherited condition resulting from mutations in mitochondrial DNA, and affected fathers do not transmit the hearing loss to their offspring. Nine loci have been reported to contribute to mitochondrial NSHL. The m.1555A>G and m.1494C>T mutations in the MT-RNR1 gene (encoding 12S rRNA) represent the most common forms of mitochondrial hearing loss, with the former being more prevalent than the latter [36]. These variants alter the secondary structure of the 12S rRNA, creating a potential binding site for aminoglycosides and thereby conferring heightened susceptibility to their ototoxic effects [36, 37]. Exposure to aminoglycosides, even at standard therapeutic doses, has been reported to induce profound, bilateral sensorineural hearing loss that may continue to progress for years after drug discontinuation [38]. Therefore, genetic screening to identify carriers, coupled with the subsequent avoidance of aminoglycosides, is crucial for preventing this form of deafness [38, 39].
Gene Therapy for NSHL
Nonsyndromic hearing impairments exhibit unparalleled heterogeneity, yet they can be generally categorized into three functional effects: loss-of-function (LOF), gain-of-function (GOF) and dominant-negative (DN) [40]. LOF variants can result in either recessive, or dominant inheritance (haploinsufficiency), where a single LOF allele is insufficient to maintain normal function. Meanwhile, GOF and DN mutations typically cause dominant disorders [41]. To counteract these mechanisms, three primary gene therapy strategies are designed: gene replacement, gene suppression, and gene editing. Accordingly, different strategies provide solutions for different pathogenic mechanisms. Gene replacement compensates for LOF through functional protein supplement, while gene suppression aims to silence or counteract the deleterious effects of GOF and DN [26]. Since gene editing enables direct intervention of DNA sequences, it holds the potential to address all variant types [41].
Advances in understanding the genetic basis of deafness, combined with improved gene delivery systems, have greatly propelled these strategies into therapy for congenital hearing loss. The first successful hearing recovery in deaf mammals was reported in 2005, using adenovirus-mediated delivery of the ATOH1 gene into the scala media of guinea pigs [42]. This approach induced hair cell regeneration and led to significant, sustained hearing recovery, particularly in high-frequency regions, despite some localized hair cell loss at the injection site [42, 43].
In 2012, VGLUT3 became the first NSHL locus successfully treated via gene replacement in mice [44]. Since then, proof-of-concept studies have been achieved for more hereditary hearing loss loci, largely focusing on recessive forms. Among the most promising therapeutic targets, such as OTOF, GJB2, USH3A, USH1C, and TMC1, the latter is the most extensively studied. Since many previous reviews have thoroughly documented the studies of deafness gene therapy during its first decade, we will not elaborate further here [26, 41, 45-47].
Gene replacement is the most widely applied strategy, frequently employing viral vectors, especially adeno-associated viruses (AAVs), due to their high transduction efficiency and sustained expression. Novel engineered AAV variants, including AAV2/Anc80L65 and AAV2/9-PHP.eB, show enhanced transduction in cochlear hair cells and are widely used in preclinical studies [48, 49].
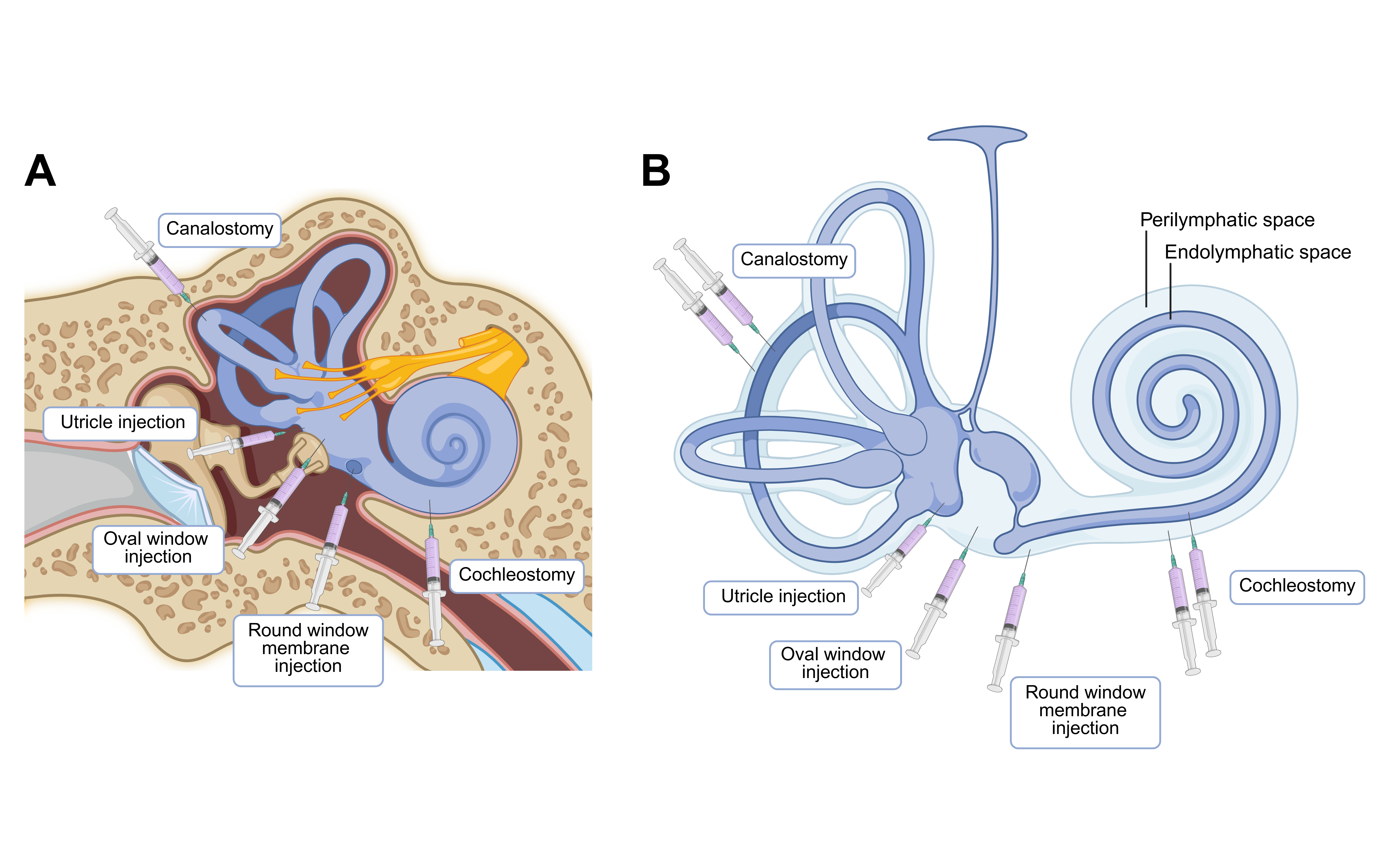
Surgical delivery methods have also evolved (Figure 1). Techniques such as round window membrane (RWM) injection, oval window delivery, utricle injection, cochleostomy, and canalostomy enable perilymphatic or endolymphatic targeting [26]. RWM injection, adapted from clinical cochlear implantation, is used in over 60% of successful cases due to its reliability and its minimal invasiveness [41]. In the landmark VGLUT3⁻/⁻ mouse study, RWM delivery achieved 100% treatment efficacy, far surpassing cochleostomy [44]; further refinements like RWM injection with canal fenestration have improved vector distribution and reduced trauma, though their safety and stability require further validation [50].
Figure 1. [PLACEHOLDER: Figure 1 legend description]
Treatment timing is another crucial factor. While interventions in mice have been applied from fetal stages to adulthood, most studies focus on the early postnatal period (P0-P10). Early intervention (P0-P2) generally yields more robust and durable hearing recovery. However, translating these timelines to humans is challenging: P10 in mice corresponds roughly to 20 weeks of human gestation, indicating that the optimal treatment window in mice aligns with the human fetal stage [26, 51-53], given the developmental lag in murine hearing. Specifically, the murine auditory system at P10 is approximately equivalent to that of a human fetus at 20 weeks of gestation [54]. Consequently, the optimal therapeutic period identified in mouse models corresponds to the human embryonic stage theoretically, which presents significant challenges for clinical translation.
Moreover, substantial anatomical and developmental differences between rodent and human inner ears limit direct clinical extrapolation [54, 55]. Non-human primate (NHP) models, with cochleae closely resembling those of humans, offer a critical translational bridge. Studies in rhesus and cynomolgus monkeys have established safe injection volumes (30-90 μL, a range relevant for human application) and demonstrated feasible AAV transduction via RWM injection, albeit with notable individual variability and dose dependency [55]. In 2022, a refined NHP RWM injection protocol, which incorporated oval window venting and transmastoid facial recess exposure, was shown to enhance delivery precision and distribution while minimizing trauma, offering a more clinically relevant approach [56].
These cumulative advances set the stage for clinical translation. A decade after the inception of inner-ear gene therapy, the first clinical trial was registered, and its first patient was enrolled at the end of 2022.
Preclinical Advances in OTOF Gene TherapyBiallelic mutations in the OTOF gene cause DFNB9, a recessive form of profound prelingual deafness classified as auditory neuropathy. The gene encodes otoferlin, a calcium-sensing protein localized in inner hair cells (IHCs) that is essential for synaptic vesicle exocytosis [29]. While structural integrity of the cochlea is preserved in DFNB9, synaptic transmission is severely impaired, leading to abnormal auditory brainstem responses but preserved otoacoustic emissions. These characteristics, combined with the relatively high prevalence of DFNB9, make OTOF an attractive target for gene therapy.
A major challenge in OTOF therapy is the gene's large coding sequence, which exceeds the packaging capacity of a single AAV vector. In early 2019, two independent studies demonstrated that dual-AAV systems could deliver full-length otoferlin cDNA into IHCs of OTOF knockout mice [57, 58].
Currently, dual-AAV gene replacement remains the dominant strategy. A team developed a dual AAV-PHP.eB system using intein-mediated trans-splicing, identifying an efficient split site that enabled stable expression of human OTOF (hOTOF) in mice and restored hearing for over 6 months [59]. Another team led by Renjie Chai developed a humanized OTOF mouse model [60]. Using an Anc80L65 vector with a hair cell-specific Myo15 promoter, they achieved full-frequency hearing recovery for at least two months in adult mice, with partial restoration lasting over 150 days. Safety and efficacy were further confirmed in cynomolgus monkeys.
Beyond auditory brainstem response (ABR) recovery, recent work by Benamer et al. demonstrated that dual AAV8-OTOF therapy also restored central auditory processing, such as frequency discrimination, in a DFNB9 mouse model, supporting the functional relevance of treatment [61]. Separately, Decibel Therapeutics (a Regeneron affiliate) characterized the expression kinetics of their candidate DB-OTO, an AAV1-based therapy using a Myo15 promoter to drive hOTOF expression [62]. In OTOF-deficient mice, OTOF mRNA and protein emerged within days and plateaued by 2-3 weeks, correlating with robust mid-frequency hearing recovery sustained for at least 3 months.
Together, these studies underscore the translational progress of OTOF gene therapy, establishing dual-AAV delivery as a viable strategy and paving the way from preclinical proof-of-concept toward clinical application.
Clinical Trials of OTOF Gene Therapy
Following promising preclinical results, a wave of clinical trials for OTOF-mediated hearing loss has commenced. To date, seven such trials are underway worldwide: four in China (ChiCTR2200063181, NCT05901480, ChiCTR2400091517, NCT06722170) and three in the USA and Europe (NCT05821959, NCT05788536, NCT06370351) (Table 3).
Table 3. OTOF gene therapies in clinical trials
| Registration date | Identifier | Drug | Serotype | Dosing strategies (per cochlear) | Injection route | Participant age | Ref. |
|---|---|---|---|---|---|---|---|
| Sep 1, 2022 | ChiCTR2200063181 | RRG-003 | AAV1 | 30/50/70/140/210μl dose-escalation groups at 2.8×1011vg/μl | Transcanal, RWM injection, OW fenestration | 6 Months and older | [12, 14, 63] |
| Feb 8, 2023 | NCT05821959 | AK-OTOF | Anc80L65 | Volume unknown; Low (Up to 4.1×1011vg) dose group; High (Up to 8.1×1011vg) dose group | Unknown | Any age | [64] |
| Mar 15, 2023 | NCT05788536 | DB-OTO | AAV1 | 240μl (7.2×1012vg) | Transmastoid facial recess, RWM injection, lateral semicircular canal fenestration | Up to 17 Years | [16, 65] |
| May 9, 2023 | NCT05901480 | OTOV101N+OTOV101C | Anc80L65 | 30μl (8.4×1011vg) to 60μl (1.68×1012vg) | Transmastoid facial recess, RWM injection | 1 Year and older | [13, 15, 66] |
| Apr 9, 2024 | NCT06370351 | SENS-501 | Unknown | Low/High dose group (not disclosed) | Unknown | 6 Months to 31 Months | [67] |
| Oct 30, 2024 | ChiCTR2400091517 | EA0010 | Unknown | Low/High dose group (not disclosed) | Unknown | 1 Year to 28 Years | [68] |
| Dec 5, 2024 | NCT06722170 | EH002 | Unknown | 25/50/100/150μl dose-escalation groups (undisclosed concentration) | Unknown | 6 Months and older | [69] |
In early 2024, the Southeast University/Otovia Therapeutics team reported initial results for their dual-vector drug OTOV101N+OTOV101C [13]. Following unilateral RWM injection via a transmastoid facial recess approach, a 5-year-old child with a contralateral cochlear implant showed rapid hearing recovery. ABR thresholds returned to normal, and pure-tone average (PTA) improved from 70-95 dB HL to 30-35 dB HL within one month, reaching near-normal levels across speech frequencies by three months. By contrast, an 8-year-old receiving bilateral injection at a higher dose exhibited more modest improvement (PTA 30-50 dB HL at 40 days), suggesting that age, dose, and individual factors may influence outcomes.
Two months later, preliminary results from the single-arm RRG-003 trial were disclosed [12]. Children aged 1-6 years showed improved auditory function and speech perception after unilateral injection, with hearing recovery emerging 4-6 weeks post-treatment. Average ABR thresholds dropped by 40-57 dB across five children. Although one 5-year-old with pre-existing AAV neutralizing antibodies showed no benefit, the overall safety profile was favorable, with no dose-limiting toxicity or serious adverse events over 26 weeks.
In 2025, the Southeast University/Otovia Therapeutics team reported updated results from a multicenter trial involving ten DFNB9 patients aged 1.5 to 23.9 years [15]. All participants showed hearing improvement over 6-12 months of follow-up, with a favorable safety profile. However, efficacy varied with age: three children aged 1-2 years responded less robustly than those aged 5-8, despite higher systemic AAV exposure. Similarly, a 14.5-year-old and a 23.9-year-old achieved only 30-45 dB PTA improvement over six months, whereas the best-responding individual improved by 87 dB. One poorly responding patient safely received a second injection in the same ear, indicating that redosing may be a viable option. These findings suggest an age-dependent therapeutic window, possibly aligned with the period of auditory cortical plasticity. Auditory deprivation during this period would perturb neural circuit maturation, whereas later intervention may fail to achieve satisfactory restoration [26, 70], explaining limited benefits in older children. In children aged 1-2 years, elevated serum neutralizing antibodies suggest a stronger immune response, potentially due to the immature blood-labyrinth barrier. The immature synaptic architecture may also inadequately support newly expressed otoferlin, resulting in decreased therapeutic efficacy. Nevertheless, the postnatal developmental trajectory of the human inner ear is not fully mapped [71], rendering such explanations hypothetical.
Most recently, Regeneron's CHORD trial evaluated DB-OTO, a dual AAV1-based gene therapy, in twelve profoundly deaf patients aged 0.9 to 16 years [16]. Participants received DB-OTO via RWM injection combined with lateral semicircular canal fenestration to facilitate perilymph drainage. By week 24, six participants could perceive soft speech and three could hear whispers. Two 16-year-olds showed partial hearing recovery, with PTA thresholds improving to 60-80 dB, consistent with earlier observations in adolescents [15]. Transient high-frequency hearing loss occurred in two patients, possibly related to injection-associated mechanical stress or perioperative infection, underscoring the need for surgical refinement and careful postoperative care.
Beyond formally published results, other gene therapies, including AK-OTOF and SENS-501, have also been reported to improve hearing safely and effectively in preliminary releases. Together, these results mark the rapid maturation of OTOF gene therapy from concept to clinically meaningful treatment for DFNB9 within just a few years.
Conclusions and prospects
Gene therapy for NSHL is advancing rapidly, with treatment of OTOF-related DFNB9 emerging as a prominent focus. The successful translation from murine models to human trials marks a historic milestone, demonstrating that congenital deafness in children can be functionally reversed. This breakthrough holds promise for improving life satisfaction and quality of life in individuals with DFNB9, while also offering a valuable framework for gene therapy targeting other forms of genetic deafness.
Nevertheless, several limitations in current findings must be addressed to achieve robust clinical translation. Although auditory function assessed via auditory brainstem response, distortion product otoacoustic emissions, and pure-tone audiometry has been consistently restored in trials, evidence remains scarce regarding recovery of higher-order auditory capacities. These include speech-in-noise understanding, music perception, and long-term language development. Furthermore, clinical research on DFNB9 is still in early stages, constrained by small patient cohorts and considerable interindividual variability, which limit statistical power. Key variables such as vector biodistribution, surgical precision, and baseline patient characteristics require further elucidation. A clearer understanding of the relationship between these factors and clinical outcomes will be essential for predicting therapeutic efficacy. Long-term safety profile of OTOF gene therapy also requires more exploration. Current studies involve only two patients with a one-year follow-up, which is inadequate to evaluate long-time risks. Therefore, larger clinical trials with extended follow-up are necessary to assess the stability of transgene expression, the durability of therapeutic benefits, and the potential immune risks associated with redosing or AAV diffusion. Finally, comparative evaluation of gene therapy versus cochlear implants awaits additional data from ongoing trials.
Despite the encouraging efficacy of OTOF-directed therapy, its overall impact may be constrained by the relatively small proportion of OTOF mutations among all hereditary hearing loss cases. Expanding the reach of deafness gene therapy will require extending research to more prevalent and genetically diverse forms of hearing loss, which in turn demands deeper mechanistic insights into underlying pathologies and inner ear physiology. Beyond biomedical challenges, social and ethical considerations, such as pediatric trial ethics, treatment affordability, and standardized diagnostic and management pathways, must also be critically addressed.
As OTOF gene therapy trials continue, effective collaboration among academic, governmental, and commercial partners will be vital to navigate these multifaceted challenges. Success in this endeavor promises to redefine the standard of care for genetic deafness, ushering in a new era of precision gene therapy for hereditary hearing loss.
Abbreviations
NSHL - Non-syndromic Hearing Loss; MET - Mechanoelectrical Transduction; LOF - Loss-of-function; GOF - Gain-of-function; DN - Dominant-negative; AAVs - Adeno-associated Viruses; RWM - Round Window Membrane; NHP - Non-human Primate; IHCs - Inner Hair Cells; hOTOF - Human OTOF; ABR - Auditory Brainstem Response; PTA - Pure-tone Average.
Declarations
Author Contributions
Yu Qi: Writing - original draft, Writing - review & editing; Fangzhi Tan: Conceptualization, Writing - review & editing, Supervision; Maoli Duan: Conceptualization, Supervision; Ling Lu: Conceptualization, Supervision, Project administration, Funding acquisition, Writing - review & editing. All authors read and approved the final manuscript.
Acknowledgement
The authors would like to thank the Clinical Genome Resource (ClinGen) for generating curated content used in this review. ClinGen's curated content was obtained from www.clinicalgenome.org.
Funding information
This work was supported by National Key R&D Program of China (2024YFC2511100/2511103) and National Natural Science Foundation of China (82471185).
Ethics Approval and Consent to Participate
Not Applicable.
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a conflict of interest at the time of conducting the study.
Data availability
This is a review article and no new data were generated or analyzed in this study.
References
[5]Walls WD, Azaiez H, Smith RJH. Hereditary Hearing Loss Homepage. Retrieved 2025.11.3.
[6]Rauschecker JP, Shannon RV. Sending Sound to the Brain. Science. 2002;295(5557):1025-1029.
[8]Macherey O, Carlyon RP. Cochlear implants. Curr Biol. 2014;24(18):R878-R884.
[25]Sheffield AM, Smith RJH. The Epidemiology of Deafness. Cold Spring Harb Perspect Med. 2019;9(9).
 Figures
Figures References
References Peer
Peer Information
InformationFigure 1. Overview of surgical delivery routes to the inner ear. (A) Anatomical locations of round window membrane injection, oval window delivery, utricle injection, cochleostomy, and canalostomy. (B) Corresponding drug distributions in perilymphatic and endolymphatic spaces of different surgical delivery routes. Created with BioRender.com.
[5]Walls WD, Azaiez H, Smith RJH. Hereditary Hearing Loss Homepage. Retrieved 2025.11.3.
[6]Rauschecker JP, Shannon RV. Sending Sound to the Brain. Science. 2002;295(5557):1025-1029.
[8]Macherey O, Carlyon RP. Cochlear implants. Curr Biol. 2014;24(18):R878-R884.
[25]Sheffield AM, Smith RJH. The Epidemiology of Deafness. Cold Spring Harb Perspect Med. 2019;9(9).
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-12-28
Accepted 2026-02-10
Published 2026-03-11